说明:分子间弱相互作用,亦称非共价相互作用或非键相互作用,是材料科学、化学等领域的核心概念之一。
本文将全面、系统地阐述分子间弱相互作用的核心概念、详细分类、理论模型和公式及其分析方法,为相关领域的研究人员提供一份内容翔实、逻辑清晰的参考资料。
水与极性溶质之间的氢键。DOI: 10.1201/9781003328209
分子间弱相互作用(Non-covalent Interactions, NCIs)是指分子与分子之间存在的、强度远低于共价键、离子键等化学键的非键合相互作用,作用能通常在 0.5~40 kJ/mol 范围内。
这类作用不涉及电子的转移或共享,主要由分子的电偶极、诱导偶极及色散效应等引发,是维系物质凝聚态结构、决定物理化学性质的核心因素。
分子间弱相互作用是一个庞大的家族,根据其物理来源和特性,可以细分为多种类型。理解每一种类型的本质对于分析和利用它们至关重要。本文主要介绍范德华力、氢键、疏水相互作用、π相互作用、卤键这几种。
范德华力是一个历史悠久且涵盖广泛的总称,最初由荷兰物理学家约翰内斯·范德华(Johannes van der Waals)为解释真实气体行为而提出。它通常包含以下三种主要的吸引力成分:取向力、诱导力和色散力。
当具有互补形状的分子靠近时发生的范德华力。DOI:10.1042/EBC20190042
取向力(Keesom Force)存在于极性分子之间。
极性分子由于内部电荷分布不均,拥有永久的电偶极矩(一端带正电,另一端带负电)。
当这些分子自由取向时,它们的偶极会倾向于异性电荷端相互靠近、同性电荷端相互远离的排列方式,从而产生净吸引力。
这种相互作用的强度与偶极矩的平方成正比,与温度成反比(高温下分子的热运动会破坏有利的取向),并且随着距离的增加以1/r6 (对于自由旋转的偶极)或 1/r3(对于固定偶极)的规律衰减。
吸引性和排斥性的偶极–偶极相互作用
诱导力(Debye Force)发生在一个极性分子和一个非极性分子之间(或者两个极性分子之间也存在)。
极性分子的永久电场会使其邻近的非极性分子的电子云发生变形,即极化,从而诱导出一个瞬时的偶极矩。这个被诱导出的偶极矩会与原有的永久偶极矩相互吸引。
诱导力的强度与极性分子的偶极矩平方以及非极性分子的极化率(polarizability)成正比,其能量同样随距离以1/r6 的规律衰减。
SAPT能量分解中的诱导项(Eind)。DOI: 10.1038/s42004-024-01329-6
色散力(London Dispersion Force)是最普遍存在的一种范德华力,即使在完全非极性的原子(如稀有气体)或分子(如甲烷、苯)之间也存在。
其来源是量子力学效应:分子中的电子在不断运动,其电子云的分布在任何瞬间都可能是不对称的,从而产生一个极其短暂的“瞬时偶极”。
这个瞬时偶极会立刻在其邻近分子中诱导出另一个与之相适应的偶极,两者随即产生吸引力。虽然单个瞬时偶极存在时间极短,但这种涨落是持续不断的,从而产生了持续的净吸引力。
瞬时偶极矩。一个氦(He)原子(a)或氢气(H?)分子(b)上瞬时偶极矩的形成,会在相邻的原子或分子中诱导出一个诱导偶极矩
色散力的大小与分子的极化率和电子数密切相关,通常分子越大、越重,电子越多,极化率越大,色散力也越强。其能量同样随距离以1/r6的规律衰减。对于大多数大分子体系,色散力往往是范德华吸引力的主要贡献者。
分子质量和表面积影响色散力的强度
氢键(Hydrogen Bond)是一种特殊且非常重要的分子间相互作用,可以看作是一种极强的偶极-偶极相互作用。
它形成的经典条件是:一个与强电负性原子(供体,Donor, D,通常是F、O、N)共价相连的氢原子(H),与另一个强电负性原子(受体,Acceptor, A,也通常是F、O、N,且该原子拥有孤对电子)之间的静电吸引。其表示为 D-H···A。
DOI:10.1042/EBC20190042
氢键的本质兼具静电相互作用、诱导、色散和少量电荷转移(共价成分)的特点。
由于H原子半径小,且其电子被D原子强烈吸引而高度裸露,使得这个带部分正电的H原子可以非常靠近受体A的孤对电子云,从而形成比普通范德华力强得多的相互作用(通常为10-40 kJ/mol)。
水分子之间的氢键。DOI: 10.1201/9781003328209
氢键具有高度的方向性(D-H···A 三原子倾向于共线)和饱和性(一个供体或受体能形成的氢键数目有限),这些特性是其在生物体系中扮演精确识别角色的基础。
此外,除了经典的N-H···O,O-H···N等强氢键外,还存在大量“非传统”或“弱”氢键,例如C-H···O/N、O-H···π等。虽然它们较弱,但在大分子体系中累积起来的效应同样不可忽视。
DNA 中的 A-T 和 C-G 碱基对中的氢键(均以虚线表示,上图);蛋白质中 β-折叠片中的氢键(左下);以及水中的氢键(中下)。在水中,氢键形成一个四面体网络(右下),其中每个水分子通过 4 个氢键相互连接。
疏水相互作用(Hydrophobic Interaction)是一个经常被误解的概念。它并非非极性分子之间存在一种特殊的吸引力,而主要是一种由溶剂(通常是水)驱动的熵效应。
A.氢键;B.疏水相互作用;C.?π-阳离子相互作 用;D.?π-π堆积相互作用;E.盐桥(离子键)。DOI:10.7506/rykxyjs1671-5187-20240914-086
当非极性分子(如油滴)被置于水中时,它们无法与水分子形成有利的氢键。为了最大化水分子之间的氢键网络,水分子会在非极性分子周围形成一个高度有序的“笼状结构”。
这种有序化导致体系的熵(混乱度)降低,在热力学上是不利的。因此,非极性分子倾向于聚集在一起,以减少它们与水接触的总表面积。
这样一来,被“释放”出来的水分子恢复了自由,体系的总熵增加,自由能降低。所以,疏水相互作用的驱动力是整个体系(溶质+溶剂)熵的增加,而非溶质分子间的直接吸引。当然,一旦非极性分子被迫靠近,它们之间的色散力也会对稳定聚集体做出贡献。
DOI:10.1016/j.biomaterials.2018.10.044
这类相互作用涉及含有π电子体系的分子,特别是芳香环,这里主要介绍π-π堆积、阳离子-π相互作用和阴离子-π相互作用。
π-π堆积(π-π Stacking)是两个或多个芳香环之间的相互作用。芳香环的π电子云在环的上下方形成负电势区域,而在环的边缘(C-H键所在处)则形成正电势区域。
因此,两个芳香环并不会像三明治一样完美地“面对面”(face-to-face)堆叠,因为这会导致π电子云之间的排斥。
更常见的稳定构象是“平行偏移”或“T形/边-面”构象,前者是两个环平行但错开,后者是一个环的边缘指向另一个环的中心,从而实现正负电势区域的互补吸引。这种相互作用在DNA碱基堆积和蛋白质内部芳香族氨基酸的排列中至关重要。
a) 展示了DNA双螺旋中碱基之间多种相互作用的共存:橙色虚线表示氢键,灰色区域表示π–π堆积相互作用
阳离子-π相互作用(Cation-π Interaction)?是一个出乎意料的强非共价相互作用,发生在阳离子和富电子的π体系之间。
阳离子的正电荷与π电子云的负电势区域之间产生强烈的静电吸引。其强度甚至可以与氢键媲美,在神经递质受体识别、离子通道选择性等生物过程中扮演关键角色。
阴离子-π相互作用(Anion-π Interaction)是阳离子-π相互作用的“镜像”,发生在阴离子和缺电子的π体系之间。为了使π体系缺电子,通常需要在芳环上连接强的吸电子基团(如-NO2, -CN,或使用含N的杂环如三嗪)。这种相互作用在阴离子识别和传感领域具有应用潜力。
卤键(Halogen Bond)是近年来备受关注的一种定向性很强的弱相互作用,类似于氢键。
当卤原子(X = Cl, Br, I)与一个电负性较强的原子共价连接时,在卤原子外侧、沿着C-X键轴的方向,会形成一个电子密度较低的区域,称为“σ-洞”(sigma-hole),这个区域带有部分正电性。
这个正电性的“σ-洞”可以与一个富电子的负电性位点(如孤对电子、π电子云或阴离子)相互吸引,形成C-X···Y(Y为路易斯碱)的相互作用,即卤键。
卤键的强度和方向性使其在晶体工程、材料设计和药物化学中成为一种非常有用的设计工具。
为了在定量层面理解和预测分子间弱相互作用,科学家们发展了一系列理论模型和计算方法,本文只介绍范德华相互作用的数学模型、静电相互作用的数学模型和氢键的特殊处理这三种。
1.范德华相互作用的数学模型:Lennard-Jones势
最广泛用于描述范德华相互作用的模型是Lennard-Jones (LJ) 12-6势,表示为
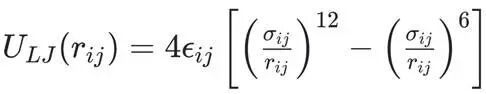
![]()
![]()
其中, rij是原子 i 和 j 之间的距离。εij是势阱深度,代表了两个原子在最优距离上相互作用的强度。σij是相互作用能为零时的原子间距,反映了原子的大小。
Lennard-Jones势
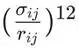
![]()
![]() 项是一个强烈的排斥项,模拟了当原子靠得太近时由电子云重叠引起的交换-排斥力。选择12次方主要是为了计算上的便利,它提供了一个足够“硬”的排斥墙。
项是一个强烈的排斥项,模拟了当原子靠得太近时由电子云重叠引起的交换-排斥力。选择12次方主要是为了计算上的便利,它提供了一个足够“硬”的排斥墙。
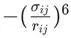
![]()
![]() 项是一个吸引项,精确地模拟了伦敦色散力的距离依赖关系 (1/r6) 。
项是一个吸引项,精确地模拟了伦敦色散力的距离依赖关系 (1/r6) 。
在实际力场中,每个原子类型都被赋予一套ε和σ参数。对于不同原子类型间的相互作用(i和j),其参数通常通过混合规则(如Lorentz-Berthelot规则)由单个原子的参数计算得到。
由排斥势和吸引势组成的弱双原子分子相互作用势的示意图
静电相互作用通常使用库仑定律来描述,模型将每个原子简化为一个携带固定部分电荷(partial charge)?的点,表达为
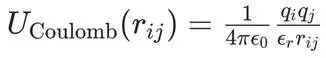
![]()
![]()
qi,qj是原子 i 和 j 上的部分电荷。这些电荷并非整数,而是通过量子化学计算(如使用RESP拟合协议)得到,以重现分子的静电势。
ε0是真空介电常数。εr是相对介电常数。在显式溶剂模型中, εr通常设为1;在隐式溶剂模型中,εr会取一个较大的值来模拟溶剂的屏蔽效应。
静电模型的成功与否,极大地依赖于部分电荷分配方案的准确性。不同的力场(如AMBER, CHARMM, OPLS)采用不同的电荷计算策略,这是它们之间差异的一个重要来源。
肽链 2I9M 在模拟中的中间结构快照:上图为使用 AMBER 力场的模拟结果,下图为采用动态极化电荷模型的模拟结果
氢键的处理方式在力场发展史上经历了重要的演变,从早期的显式氢键项到现代的隐式处理。
早期的力场,如AMBER的早期版本(1984年),引入了一个专门的函数来描述氢键,最常见的是10-12势,表示为
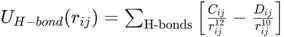
![]()
![]()
这个函数形式类似于L-J势,但吸引项使用了1/r10得其作用范围比色散力更短,更能体现氢键的局域性。通常还会乘以一个角度依赖项来描述氢键的方向性。
取向依赖的氢键能量函数
现代主流力场,如AMBER94及之后版本、CHARMM和OPLS,不再包含显式的氢键项。它们认为氢键的物理本质(主要是静电,辅以诱导和色散)已经可以被库仑项和L-J项的组合很好地描述 。
通过对氢键供体和受体原子(特别是与氢相连的重原子和氢原子本身)的部分电荷和L-J参数进行精细的优化,使得标准的非键模型能够自动、准确地再现氢键的能量、距离和方向性。
这种隐式处理方式更加物理自洽,且避免了预先定义哪些原子对可以形成氢键的麻烦,使得模型更具普适性和可转移性。
力场中描述的相互作用
要全面地研究分子间弱相互作用,需要结合实验测量和理论计算。实验提供了真实世界的数据,而计算则提供了原子尺度的细节和物理本质的解释。
光谱学方法主要通过监测分子间相互作用对分子能级(振动、转动、电子、核自旋能级)的影响来研究弱相互作用,尤其适用于溶液相的研究。
1.光谱(IR):氢键会使 O-H、N-H 等伸缩振动峰向低波数位移,峰形宽化;π-π 堆积作用会导致芳香环的特征吸收峰强度和位置变化。
2.核磁共振(NMR):分子间弱相互作用会影响原子核的化学位移,例如氢键使 1H NMR 的化学位移向低场移动;通过 NOESY 谱可分析分子间的空间邻近关系,表征 π-π 堆积、卤键等作用。
3.紫外 - 可见吸收光谱(UV-Vis):π-π 堆积作用会使共轭分子的吸收峰红移或蓝移,吸收强度发生变化。
UV-Vis图。DOI:10.1002/chem.202404000
1.X 射线单晶衍射(XRD):X射线单晶衍射是确定分子在晶体中三维结构的“金标准”,能够精准测定分子间的键长、键角,直接表征氢键、卤键、π-π 堆积的存在及构型。通过晶体结构解析,可明确弱相互作用在晶体堆积中的连接方式。
2.粉末 X 射线衍射:用于分析多晶样品中分子的堆积模式,间接反映弱相互作用的类型。
单晶XRD配备四圆测角仪,用于高纯度单晶结构解析。DOI:10.1080/10408398.2024.2395487
1.密度泛函理论(DFT):计算分子的电荷分布、偶极矩、极化率,预测弱相互作用的存在及强度;通过能量分解分析(EDA),定量区分范德华力、氢键等不同作用的贡献占比。
2.分子动力学模拟(MD):模拟凝聚态体系中分子的运动轨迹,分析弱相互作用的动态变化,揭示其对材料宏观性能的影响机制。
DFT计算。DOI:10.1021/jacs.4c15280
差示扫描量热法(DSC):通过测定物质相变过程的焓变,间接反映分子间弱相互作用的强度;例如,分子间氢键越强,晶体的熔融焓越高。
分子间弱相互作用是连接微观分子世界与宏观物质现象的关键桥梁。它们虽然“弱”,但其多样性、协同性、方向性和动态性,赋予了物质世界以结构、功能和生命力。展望未来,随着实验技术的精度不断提升,计算能力的指数级增长,我们对分子间弱相互作用的认知和调控能力必将迈上一个新的台阶。
文章来源:
微信公众号—材料有干货
声明:转载此文是出于传递更多信息之目的。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢